Gece körlüğü
| Gece körlüğü | |
|---|---|
 | |
| Uzmanlık | Oftalmoloji |
Retinitis Pigmentosa (RP)
Retinitis pigmentosa (RP), halk arasında tavuk karası ve gece körlüğü adlarıyla bilinen ve görme kaybına neden olan genetik bir göz hastalığıdır[1]. Her 4.000 kişide 1'i etkilediği tahmin edilmektedir[1].
Belirtileri arasında gece görmede güçlük, azalan periferik görüş (yanal ve üst veya alt görsel alan), çevresel (periferik) görüşte azalma ile gelebilen "tünel görüşü" mevcuttur[1]. Tam körlük nadirdir[2]. Belirtilerin başlangıcı çoğunlukla yavaştır ve genellikle çocuklukta başlar.[1][2]
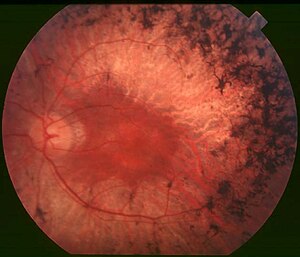
Retinitis pigmentosa genellikle bir veya her iki ebeveynden kalıtsal olarak geçer[3]. Neredeyse 100 gendeki genetik varyantlar tarafından oluşturulur[3]. Hastalığın oluşma mekanizması, göz küresinin retinasını kaplayan çubuk fotoreseptör hücrelerinin kademeli olarak kaybını içerir[1]. Çubuk hücreleri, koni hücrelerini hücre ölümünden (apoptoz) koruyan nöroprotektif bir madde (Çubuk kaynaklı koni canlılık faktörü, RdCVF) salgılar. Ancak çubuk hücreleri öldüğünde, bu madde artık sağlanmaz. Bu durumu genellikle koni fotoreseptör hücrelerinin ölümü izler[1]. Teşhis, retinaya göz muayenesi ile yapılır. Bu muayenede, altta yatan retinal pigment epitel (RPE) hücrelerinin yırtılmasıyla oluşan koyu pigment birikintileri görülür. Çünkü bu hücreler melanin adı verilen bir pigment içerir.[1] Diğer destekleyici testler elektroretinogram (ERG), görme alanı testi (VFT), optik koherens tomografi (OCT) ve kişinin belirli RP tipinden sorumlu geni belirlemek için DNA testi içerebilir.[1]
Retinitis pigmentosa için şu anda bir tedavi yoktur.[2] Görmede yaşanan sorunları yönetme çabaları; büyüteç gibi düşük görme yardımcıları, taşınabilir aydınlatma, oryantasyon ve mobilite eğitimi kullanımı gibi yöntemler içerebilir.[1] A vitamini (Retinil) palmitat takviyeleri kötüleşmeyi yavaşlatmada faydalı olabilir.[1] Ciddi vakaları olan bazı kişiler için yapay retina gibi protezler bir seçenek olabilir.[1]
Şu anda ticari olarak yalnızca tip 2 LCA sahibi RP hastalarına sunulan bir FDA onaylı gen tedavisi vardır. Bu tedavi, retinal pigment epitel (RPE) hücreleri içinde üretilen hatalı kodlanmış RPE65 proteinini değiştirir. Tedaviyi alan hastaların yaklaşık %50'sinde etkili bir şekilde çalıştığı görülmüştür. Hasta, tercihen çocukluk döneminde, RPE65 tedavisini ne kadar erken alırsa olumlu sonuç alma şansı o kadar artar. Şu anda araştırılan birçok başka tedavi vardır ve bunların önümüzdeki birkaç yıl içinde onaylanması hedeflenmektedir.
Belirti ve Semptomlar
Retinitis pigmentosanın başlangıç semptomları, azalmış gece görüşü (niktalopi) ve orta-çevresel görüş alanının kaybı ile tanınmaktadır[4]. Düşük ışıkta görme yeteneğinden sorumlu ve çoğunlukla retinal periferde bulunan çubuk (rod) fotoreseptör hücreleri, bu hastalığın non-sendromik (başka sağlık sorunları olmayan) formlarında ilk etkilenen retinal yapılardır.[5] Görme kaybı, çevresel görüş alanından merkeze doğru ilerler ve sonunda tünel görüşü arttıkça merkezi görüş alanına kadar uzanır. Çubuk fotoreseptör hücrelerinin kaybına eşlik edecek şekilde koni fotoreseptör hücrelerinin kaybı, bu nedenle de renkli görme ve görüş keskinliği bozulabilir[5]. Hastalığın seyri genelde her iki gözde benzer şekilde ancak birebir aynı olmayacak şekilde gerçekleşir. Retinitis pigmentosa, başlangıçtaki çubuk fotoreseptör dejenerasyonunun ve daha sonraki koni fotoreseptör kaybının etkileriyle birlikte çeşitli dolaylı semptomlarla tanımlanmaktadır. Normal seviyelerdeki ışığın yoğun bir parlama olarak algılandığı fotofobi ve görüş alanında yanıp sönen, dönen veya parıldayan ışıklar görülen fotopsi gibi olgular genellikle RP'nin daha ileri aşamalarında ortaya çıkar.
Non-sendromik RP (RP başka eşlik eden hastalıklar olmadan tek başına ortaya çıkar) genellikle aşağıdaki semptomlardan birçoğunu gösterir:
- Gece körlüğü
- Tünel görüş (çevresel görüş kaybına bağlı)
- Derinlik algısı kaybı (perspektif algısında bozulma)[6]
- Fotopsi (Görüş alanında yanıp sönen/titreşen/dönen/parıldayan ışıklar oluşması)
- Fotofobi (Parlak ışığa karşı aşırı hassasiyet)
- Karanlık ortamlardan aydınlık ortamlara ve tersi geçişlerde yavaş adapte olma
- Görme bulanıklığı
- Zayıf renk ayrımı
- Merkezi görme kaybı en son meydana gelir, çünkü bu bir çubuk fotoreseptör hastalığıdır ve merkezi görme bölgesinde en çok bulunan koni fotoreseptörleri daha az etkiler.
- Resmi körlük. Hastaların çoğu tamamen kör olmaz ve genellikle sınırlı veya işlevsel olmayan görme yeteneğini korur.
Sebepler
Retinitis Pigmentosa üç şekilde ortaya çıkabilir[7]:
- Tek başına (Non-sendromik): RP başka herhangi bir klinik bulgu olmadan tek başına ortaya çıkar.
- Diğer rahatsızlıklarla birlikte (Sendromik): RP; işitme kaybı, nörolojik bozukluklar, gelişimsel anomaliler veya başka karmaşık klinik bulgular ile birlikte görülebilir.
- Diğer sistemik hastalıklara bağlı (Sekonder): RP, başka bir sistemik hastalığın bir sonucu olarak ortaya çıkabilir.
RP ile birlikte görülebilen bazı hastalıklar:
- Usher sendromu: RP ve doğuştan sağırlık veya ilerleyici sağırlığın birlikte görülmesidir.[8]
- Alport sendromu: RP, böbrek sorunlarına yol açan anormal bir glomerüler bazal membran sebepli oluşan nefrotik sendrom ile birlikte görülür. X’e bağlı dominant olarak kalıtılır.
- Kearns-Sayre sendromu (KSS): RP; göz hareketlerinde kısıtlılık (oftalmopleji), göz kapağı düşüklüğü ve kardiyak ileti sisteminde bozukluklar ile birlikte görülür. Mitokondriyal DNA silinmesine (delesyon) bağlı bir bozukluktur.
- Abetalipoproteinemi: RP; yağ ve yağda eriyen vitaminlerin (A,D,E,K) emiliminde bozukluk, denge ve hareket bozukluğu içeren ataksi, steatore (yağlı dışkı) ve akantoz (yıldız şekilli alyuvarlar) ile birlikte görülür.[9]
- McLeod sendromu: RP, kas distrofisi ve kronik granulomatoz hastalığı gibi diğer nadir genetik hastalıklarla birlikte görülebilir. X’e bağlı resesif kalıtılan bir hastalıktır.
- Bardet-Biedl sendromu: RP, hipogonadizm (testis ve yumurtalıklarda hormon üretim bozukluğu) ve gelişimsel gecikme ile birlikte görülür. Otozomal resesif olarak kalıtılır.[10]
- Diğer bazı durumlar nörosifiliz, toksoplazmoz ve Refsum hastalığı’dır.
RP’ye benzer oftalmoskopik bulgulara sahip bazı kazanılmış hastalıkların sebepleri arasında erken yaşta geçirilen enfeksiyonlar ile ilişkili göz iltihabı, otoimmün paraneoplastik retinopati, ilaç toksisitesi ve göz travması bulunmaktadır. Bu hastalıklar unilateral (tek gözde) veya bilateral (her iki gözde) olabilir, ilerlemesi durabilir veya devam edebilir.[11][12]
Genetik
Retinitis Pigmentosa (RP) kalıtsal retina bozukluklarının (IRD) en yaygın şekillerinden biridir.[13] Görsel yolda ihtiyaç duyulan proteinleri kodlayan pek çok gen vardır ve bu genlerdeki mutasyonlar RP fenotipine neden olabilir.[14] RP'nin kalıtım biçimleri otozomal dominant, otozomal resesif, X’e bağlı ve maternal (mitokondriyal) olarak tanımlanmıştır ve bunlar ebeveyn neslindeki spesifik gen mutasyonlarına bağlıdır.
1989 yılında, düşük ışık koşullarında görmeyi sağlayan görsel transdüksiyon çağlayanında (cascade) önemli bir rol oynayan bir pigment olan rodopsin geni için bir mutasyon tanımlanmıştır. Rodopsin geni, fotoreseptör dış segmentlerinin başlıca bir proteinini kodlar. Bu gendeki mutasyonlar en sık olarak anlamsız (nonsense) mutasyonlardır. Bazı durumlar ise rodopsin proteininin yanlış katlanması sonucu görülür. Rodopsin geninin keşfinden bu yana, tüm retina dejenerasyonu tiplerinin %15'ini ve otozomal dominant RP formlarının yaklaşık %25'ini oluşturan 100'den fazla RHO mutasyonu tanımlanmıştır.[13][15]
RP'nin otozomal resesif kalıtım biçimleri en az 45 gende tanımlanmıştır.[16] Bu, aynı RP'yi tetikleyen gen mutasyonunun diallelik formunda taşıyıcı olan iki etkilenmemiş bireyin RP fenotipli çocukları olabileceği anlamına gelir. USH2A genindeki bir mutasyonun, otozomal resesif biçimde kalıtıldığında Usher Sendromu olarak bilinen sendromik bir RP'nin %10-15'ine neden olduğu bilinmektedir.[17]
Dört pre-mRNA splice faktöründeki mutasyonların otozomal dominant RP’ye neden olduğu bilinmektedir. Bunlar PRPF3 (insan PRPF3 HPRPF3'tür; ayrıca PRP3), PRPF8, PRPF31 ve PAP1'dir. Bu faktörler her yerde ifade edilir ve evrensel bir faktördeki (her yerde ifade edilen bir protein) kusurların sadece retinada hastalığa neden olacağı öne sürülmektedir, çünkü retina fotoreseptör hücreleri diğer hücre tiplerine göre çok daha fazla protein (rodopsin) işleme ihtiyacı duyar.[18]
RP'nin somatik veya X’e bağlı kalıtım biçimleri şu anda altı genin mutasyonları ile tanımlanmaktadır, en yaygın olarak RPGR ve RP2 genlerindeki belirli lokuslarda meydana gelmektedir.[17]
Tanı
Retinitis pigmentosa için doğru tanı, fotoreseptör hücre fonksiyonunun ilerleyici kaybının belgelenmesine dayanır. Görme alanı ve görme keskinliği testleri, optik koherens tomografi (OCT) ve elektroretinografi (ERG) ile birlikte hastalığın varlığı doğrulanır.[19]
Görme alanı ve keskinliği testleri, hastanın görme alanının büyüklüğünü ve görsel algısının netliğini sağlıklı 20/20 görme ile karşılaştırır. RP’ye işaret eden tanısal özellikler, görme alanı testinde küçük ve giderek azalan bir görme alanı ve görme keskinliği testi sırasında ölçülen netlikteki bozulmadır.[20] Fundus ve retinal (optik koherens) görüntüleme gibi optik tomografi teknikleri RP tanısı koyarken kullanılabilir. Fotoreseptör kalınlığı, retina tabakası morfolojisi ve retinal pigment epitel fizyolojisi hakkında fikir veren optik koherens tomografisi ile kesit görüntüleme ve bununla birlikte fundus görüntüleme; RP’nin ilerleyiş durumunu belirlemeye yardımcı olabilir.[21]
Görme alanı ve keskinliği testi sonuçları, retinal görüntüleme ile birlikte retinitis pigmentosa tanısını desteklemektedir. Ancak hastalığın diğer patolojik özelliklerini doğrulamak için ek testler gereklidir. Elektroretinografi (ERG), fotoreseptör dejenerasyonunun işlevsel yönlerini değerlendirerek RP tanısını doğrular ve semptomların ortaya çıkmasından önce fizyolojik anormallikleri tespit edebilir. Farklı hızlardaki ışık darbelerine fotoreseptör hücrelerin yanıtı ölçülürken göz üzerine bir elektrot lens uygulanır. RP fenotipini gösteren hastalar, çubuk fotoreseptörlerde azalmış veya gecikmiş elektriksel yanıtın yanı sıra muhtemelen bozulmuş koni fotoreseptör hücre yanıtı gösterirler.
Retinitis pigmentosanın genetik kalıtım şekli nedeniyle hastanın aile öyküsü de tanı koymada dikkate alınır. En az 35 farklı gen veya lokusun sendromik olmayan RP'ye (başka bir hastalığın sonucu olmayan RP) neden olduğu bilinmektedir. RP mutasyon tipi hakkında bilgiler, klinik olarak aşağıdaki genler için mevcut olan DNA testi ile belirlenebilir:
- RLBP1 (otozomal resesif, Bothnia tipi RP)
- RP1 (otozomal dominant, RP1)
- RHO (otozomal dominant, RP4)
- RDS (otozomal dominant, RP7)
- PRPF8 (otozomal dominant, RP13)
- PRPF3 (otozomal dominant, RP18)
- CRB1 (otozomal resesif, RP12)
- ABCA4 (otozomal resesif, RP19)
- RPE65 (otozomal resesif, RP20)[22]
Diğer tüm genler (örneğin DHDDS) için moleküler genetik testler henüz sadece araştırma amaçlı olarak mevcuttur.
Tedavi
Retinitis pigmentosa için şu anda bir tedavi bulunmamaktadır, ancak çeşitli tedavi olasılıklarının etkinliği ve güvenliği değerlendirilmektedir. Vitamin A, DHA, NAC ve lutein gibi çeşitli takviyelerin hastalığın ilerlemesini geciktirmedeki etkinliği belirsizliğini korumakta, ancak umut verici bir tedavi seçeneği olarak değerlendirilmektedir.[13][23] Optik protez cihazları, gen terapisi ve retina tabakası nakillerini araştıran klinik denemeler, RP hastalarında görmenin kısmen geri kazandırılması konusundaki aktif çalışma alanlarıdır.[24]
Hastalığın Geciktirilmesi
Çalışmalar, günlük 15.000 IU (4.5 mg eşdeğeri) vitamin A palmitat alımının çubuk fotoreseptör dejenerasyonunu geciktirdiğini göstermiştir; böylece bazı hastalarda hastalığın ilerlemesi durdurulmaktadır.[25] Son araştırmalar, uygun vitamin A takviyesinin bazı hastalarda hastalığın belirli evrelerinde körlüğü 10 yıla kadar erteleyebileceğini (yıllık %10 kaybı %8.3'e düşürerek) göstermiştir.[26]
Kemik İliği Kökenli Kök Hücreler (BMSC)
Retina ve optik sinir hastalıklarının tedavisinde otolog kemik iliği kökenli kök hücreleri (BMSC) kullanan bir klinik araştırma şirketi olan MD Stem Cells, NIH kayıtlı devam eden Kök Hücre Oftalmoloji Çalışması II (SCOTS2) klinik denemesi (NCT 03011541) kapsamında Retinitis Pigmentosa kohortundan sonuçlar yayımladı.[27] Sonuçlar umut vericiydi; gözlerin %45.5'i ortalama 7.9 satır iyileşme gösterdi (temele göre %40.9 LogMAR iyileşmesi) ve gözlerin %45.5'i takip süresince stabil keskinlik gösterdi. Sonuçlar istatistiksel olarak anlamlıydı (p=0.016). Retinitis Pigmentosa, bu çalışmada tedavi edilmeye ve değerlendirilmeye devam edilmektedir.[28]
Argus Retina Protezi
Argus retina protezi, Şubat 2011'de hastalık için onaylanan ilk tedavi oldu ve şu anda Almanya, Fransa, İtalya ve Birleşik Krallık'ta mevcuttur[29]. 30 hasta üzerinde yapılan uzun dönem denemelerin ara sonuçları 2012'de yayımlandı.[30] Argus II retina implantı, ABD'de de piyasa onayı almıştır.[31] Cihaz, şekil ve hareket algılama yetisini kaybetmiş RP'li yetişkinlerin daha hareketli olmasına ve günlük aktiviteleri gerçekleştirmesine yardımcı olabilir. Haziran 2013'te, ABD'deki on iki hastane, Argus II'nin lansmanına hazırlık olarak RP'li hastalar için yakında danışmanlık kabul edeceklerini duyurdu.[32] Alpha-IMS, optik fovea altına küçük bir görüntü kaydedici çipin cerrahi olarak yerleştirilmesini içeren bir retina altı implanttır. Alpha-IMS çalışmalarından elde edilen görsel iyileşme ölçümleri, klinik denemelere geçilmeden ve piyasa onayı verilmeden önce cihazın güvenliğinin gösterilmesini gerektirir.[33]
Gen Terapisi
Gen terapisi çalışmalarının amacı, retinitis pigmentosa fenotipi ile ilişkili mutant genleri ifade eden retina hücrelerini sağlıklı gen formları ile viral olarak takviye etmektir; böylece, yerleştirilen sağlıklı genin sağladığı talimatlar doğrultusunda retina fotoreseptör hücrelerinin onarımı ve doğru çalışması sağlanır. LCA2 retinitis pigmentosa fenotipini ifade eden retinaya sağlıklı RPE65 geninin yerleştirilmesini araştıran klinik denemeler, görmede oluşan iyileşmeleri ölçtü; ancak, retina fotoreseptörlerinin dejenerasyonu hastalıkla ilgili hızda devam etti[34]. Gen terapisi, kalan sağlıklı retina hücrelerini korurken, zaten hastalıklı olan fotoreseptör hücrelerinde daha önce biriken hasarı onaramayabilir.[24] Gen terapisi, teorik olarak, fotoreseptör düşüşünün en kısa ilerlemesini sergileyen genç hastalara fayda sağlayabilir; böylece, yerleştirilen sağlıklı gen ile hücre kurtarma olasılığı daha yüksek olur.[35]
İlaçlar
UC Berkeley'de yapılan bir çalışmada, insanlarda alkolikliği tedavi etmek için kullanılan disülfiram adlı ilacın, retinitis pigmentosalı sıçanlarda, hastalığın geç evrelerinde bile görme kaybını kısmen geri kazandırma potansiyeline sahip olduğu bulundu.[36][37][38] İnsanlarda bu araştırmanın devam etmesi için çalışmalar sürdürülmektedir.
Gidişat
Retinitis pigmentosa'nın kesin bir tedavisinin olmaması, bu hastalığa sahip hastalar için kaçınılmaz olarak cesaret kırıcı bir görünüm oluşturur. Tam körlük nadir olmakla birlikte, kişinin görme keskinliği ve görme alanı, başlangıçta çubuk fotoreseptörlerin ve daha sonra koni fotoreseptörlerin dejenerasyonu ilerledikçe azalmaya devam edecektir.[4]
Çalışmalar, hastalık genotipini taşıyan çocukların, ilerleyici görme kaybı ile ilişkili fiziksel ve sosyal etkiler için hazırlanmak amacıyla semptom öncesi danışmanlıktan fayda sağladığını göstermektedir. Aktif danışmanlık[39] ile psikolojik prognoz hafifletilebilse de, hastalığın fiziksel etkileri ve ilerlemesi büyük ölçüde başlangıç semptomlarının ortaya çıkma yaşına ve fotoreseptör dejenerasyonunun hızına bağlıdır, olası tedavilere erişime değil. Düşük Görme Uzmanları tarafından sağlanan düzeltici görsel yardımcılar ve kişiselleştirilmiş görme terapisi, hastaların görme keskinliğindeki hafif bozuklukları düzeltmelerine ve kalan görme alanlarını optimize etmelerine yardımcı olabilir. Destek grupları, görme sigortası ve yaşam tarzı terapisi, ilerleyici görme kaybını yönetenler için ek faydalı araçlardır.[19]
Epidemiyoloji
Retinitis pigmentosa, kalıtsal körlüğün başlıca nedenidir ve yaklaşık 1/4000 kişi yaşamları boyunca hastalığın non-sendromik formunu deneyimlemektedir[40][41]. Dünya genelinde tahminen 1,5 milyon insan şu anda etkilenmiş durumdadır. Erken başlangıçlı RP, yaşamın ilk birkaç yılı içinde ortaya çıkar ve tipik olarak sendromik hastalık formları ile ilişkilidir, geç başlangıçlı RP ise erken yetişkinlikten orta yaşa kadar ortaya çıkar.
Otozomal dominant ve resesif formlar hem erkek hem de kadın popülasyonlarını eşit şekilde etkiler; ancak daha az yaygın olan X bağlantılı form, X bağlantılı mutasyonun erkek alıcılarını etkilerken, kadınlar genellikle RP özelliğinin taşıyıcıları olarak etkilenmezler. X bağlantılı hastalık formları genellikle şiddetlidir ve ilerleyen evrelerde tam körlüğe yol açar. Nadir durumlarda, X bağlantılı gen mutasyonunun dominant bir formu hem erkekleri hem de kadınları eşit şekilde etkiler.[42]
RP'nin genetik kalıtım kalıpları nedeniyle, birçok izole popülasyonda daha yüksek hastalık sıklıkları veya belirli bir RP mutasyonunun artmış prevalansı görülmektedir. Retinitis pigmentosa'da çubuk fotoreseptör dejenerasyonuna katkıda bulunan önceden var olan veya yeni ortaya çıkan mutasyonlar aile hatları boyunca aktarılır; böylece belirli RP vakalarının, hastalığın soy geçmişine sahip belirli coğrafi bölgelere yoğunlaşmasına izin verir. Maine (ABD), Birmingham (İngiltere), İsviçre (1/7000), Danimarka (1/2500) ve Norveç'teki farklı prevalans oranlarını belirlemek için çeşitli kalıtsal çalışmalar yapılmıştır.[43] Navajo Kızılderilileri de yüksek RP kalıtım oranı sergiler, bu oran tahminen 1878 kişide 1 olarak belirlenmiştir. Belirli aile hatları içindeki RP sıklığının artmasına rağmen, hastalık ayrımcı olmayan bir şekilde dünya genelindeki tüm popülasyonları eşit şekilde etkiler.
Araştırmalar
Gelecekteki tedaviler retina nakilleri[44], yapay retina implantları[45], gen terapisi, kök hücreler, besin takviyeleri ve/veya ilaç tedavilerini içerebilir. 2012 yılında, Miami Üniversitesi Bascom Palmer Göz Enstitüsü'ndeki bilim insanları, mesensefalik astrosit kaynaklı nörotrofik faktör (MANF) enjekte edilen hayvan modelinde fotoreseptörlerin korunduğunu gösteren veriler sundular.[46][47] Kaliforniya Üniversitesi, Berkeley'deki araştırmacılar, hasar görmüş çubuk ve koni hücrelerine sahip hayvanlarda retinal ganglion hücrelerini aktive eden bir "fotoanahtar" kullanarak kör farelerde görmeyi geri kazandırmayı başardılar.[48] 2015 yılında, Cedars-Sinai Tıp Merkezi'nde Bakondi ve ekibi tarafından yapılan bir çalışma, CRISPR/Cas9'un otozomal dominant retinitis pigmentosa formuna sahip sıçanları tedavi etmek için kullanılabileceğini gösterdi.[49] Araştırmacılar, rod kaynaklı koni yaşam faktörü (RdCVF) ve Nrf2'nin retinitis pigmentosa fare modellerinde koni fotoreseptörlerini koruyabileceğini buldular.[50][51] 2016 yılında, RetroSense Therapeutics, ışığa duyarlı alglerden DNA içeren virüsleri retinitis pigmentosa'lı birkaç kör insanın gözlerine enjekte etmeyi amaçladı. Başarılı olursa, siyah beyaz görmeleri mümkün olacaktı.[52][53] 2017 yılında FDA, biallelik RPE65 mutasyonuyla ilişkili retinal distrofiyi tedavi etmek için gen terapisi voretigene neparvovec'i onayladı. 2020 yılında, bir literatür incelemesi, o zamanki kanıtlara dayanarak, deneysel bir terapötik teknik olan transkorneal elektriksel stimülasyonu retinitis pigmentosa'da "muhtemelen etkili" (seviye B) olarak değerlendirdi.[52] 2021 yılında, Channelrhodopsin proteininin optogenetik bir uygulaması, sadece bir hastadan oluşan bir seride işlevsel olmayan görmenin kısmi olarak geri kazanıldığını bildirdi. Standart protokolü kullanmadılar, ancak kendi kriterlerini oluşturdular.[54] Kullanılan yeni algal channelrhodopsin, 1000 Bitki Genom Projesi'nden rastlantısal olarak keşfedildi.[55]
Bilindik Vakalar
- Jennifer L. Armentrout, Amerikalı yazar
- Walt Bodine, Amerikalı yayıncı,
- Willie Brown, San Francisco, California'nın 41. belediye başkanı
- Alex Bulmer, Kanadalı oyun yazarı[56]
- Molly Burke, Kanadalı YouTuber ve motivasyon konuşmacısı
- Mark Erelli, Amerikalı şarkıcı/şarkı yazarı, Lori McKenna'nın gitarist[57]
- Neil Fachie, Britanyalı paralimpik bisikletçi[58]
- William (Bill) Fulton, Kent plancısı, yazar ve eski Ventura, California belediye başkanı
- Gordon Gund, Amerikalı iş insanı ve profesyonel spor takımı sahibi
- Rigo Tovar, Meksikalı müzisyen, şarkıcı ve aktör
- Lindy Hou, Avustralyalı tandem bisikletçi ve triatlet[59]
- Amar Latif, Girişimci, televizyon kişiliği ve profesyonel gezgin
- Rachael Leahcar, Avustralyalı şarkıcı/şarkı yazarı, aktris ve motivasyon konuşmacısı
- Steve Lonegan, Bogota, New Jersey belediye başkanı; ABD Senatosu için Cumhuriyetçi aday[60]
- Chris McCausland, Britanyalı stand-up komedyeni ve aktör
- Robin Millar, İngiliz plak yapımcısı, müzisyen ve iş insanı[61]
- Woody Shaw, Amerikalı caz trompetçisi[62]
- Regina Sorenson, Avustralyalı televizyon kişiliği[63]
- Shel Talmy, Amerikalı plak yapımcısı, şarkı yazarı ve düzenleyici[64]
- Sabriye Tenberken, Alman Tibetolog ve Tibet Braille alfabesi geliştiricisi
- Danelle Umstead, Amerikalı Paralimpik alp disiplini kayakçısı, Dancing with the Stars yarışmacısı[65]
- Jon Wellner, Amerikalı aktör[66]
- Steve Wynn, Amerikalı iş adamı ve Las Vegas casino geliştiricisi[67]
- Sheena Iyengar, Columbia Business School'da Yönetim Bölümü'nde S.T. Lee İşletme Profesörü[68]
Ayrıca bakınız
Kaynakça
- ^ a b c d e f g h i j k "Facts About Retinitis Pigmentosa | National Eye Institute". web.archive.org. 7 Mart 2019. Erişim tarihi: 26 Temmuz 2024.
- ^ a b c https://web.archive.org/web/20170829205230/http://kellogg.umich.edu/patientcare/downloads/Understand-Retinitis-Pigmentosa.pdf
- ^ a b "Entry - #268000 - RETINITIS PIGMENTOSA; RP - OMIM". www.omim.org. Erişim tarihi: 26 Temmuz 2024.
- ^ a b Shintani, Kelly; Shechtman, Diana L.; Gurwood, Andrew S. (1 Temmuz 2009). "Review and update: Current treatment trends for patients with retinitis pigmentosa". Optometry - Journal of the American Optometric Association. 80 (7): 384–401. doi:10.1016/j.optm.2008.01.026. ISSN 1529-1839.
- ^ a b Soucy, Ed; Wang, Yanshu; Nirenberg, Sheila; Nathans, Jeremy; Meister, Markus (1998-09). "A Novel Signaling Pathway from Rod Photoreceptors to Ganglion Cells in Mammalian Retina". Neuron. 21 (3): 481–493. doi:10.1016/s0896-6273(00)80560-7. ISSN 0896-6273. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ Prem Senthil, M; Khadka, J; Pesudovs, K (2017-05). "Seeing through their eyes: lived experiences of people with retinitis pigmentosa". Eye. 31 (5): 741–748. doi:10.1038/eye.2016.315. ISSN 0950-222X. PMC 5437327 $2. PMID 28085147. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ Daiger, SP; Sullivan, LS; Bowne, SJ (2013-08). "Genes and mutations causing retinitis pigmentosa". Clinical genetics. 84 (2): 10.1111/cge.12203. doi:10.1111/cge.12203. ISSN 0009-9163. PMC 3856531 $2. PMID 23701314. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ "Usher syndrome". Retina UK (İngilizce). Erişim tarihi: 26 Temmuz 2024.
- ^ "Types of Mitochondrial Myopathies (MM) - Diseases". Muscular Dystrophy Association (İngilizce). 18 Aralık 2015. Erişim tarihi: 26 Temmuz 2024.
- ^ "Bardet-Biedl syndrome (BBS)". Retina UK (İngilizce). 6 Eylül 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 26 Temmuz 2024.
- ^ Adamus, Grazyna; Ren, Gaoying; Weleber, Richard G. (4 Haziran 2004). "Autoantibodies against retinal proteins in paraneoplastic and autoimmune retinopathy". BMC Ophthalmology. 4 (1): 5. doi:10.1186/1471-2415-4-5. ISSN 1471-2415. PMC 446200 $2. PMID 15180904. KB1 bakım: PMC biçimi (link)
- ^ Bastek, James V.; Foos, R.Y.; Heckenlively, John (1981-11). "Traumatic Pigmentary Retinopathy". American Journal of Ophthalmology (İngilizce). 92 (5): 621–624. doi:10.1016/S0002-9394(14)74652-5. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ a b c Hartong, Dyonne T; Berson, Eliot L; Dryja, Thaddeus P (2006-11). "Retinitis pigmentosa". The Lancet. 368 (9549): 1795–1809. doi:10.1016/s0140-6736(06)69740-7. ISSN 0140-6736. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ "Entry - #268000 - RETINITIS PIGMENTOSA; RP - OMIM". omim.org. Erişim tarihi: 26 Temmuz 2024.
- ^ jamanetwork.com. doi:10.1001/archopht.1991.01080010094039 https://jamanetwork.com/journals/jamaophthalmology/fullarticle/638853. Erişim tarihi: 26 Temmuz 2024. Eksik ya da boş
|başlık=(yardım) - ^ Rivolta, C. (15 Mayıs 2002). "Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns". Human Molecular Genetics. 11 (10): 1219–1227. doi:10.1093/hmg/11.10.1219. ISSN 1460-2083.
- ^ a b "Retinitis pigmentosa: MedlinePlus Genetics". medlineplus.gov (İngilizce). Erişim tarihi: 26 Temmuz 2024.
- ^ Bujakowska, Kinga; Maubaret, Cecilia; Chakarova, Christina F.; Tanimoto, Naoyuki; Beck, Susanne C.; Fahl, Edda; Humphries, Marian M.; Kenna, Paul F.; Makarov, Evgeny; Makarova, Olga; Paquet-Durand, François (1 Aralık 2009). "Study of Gene-Targeted Mouse Models of Splicing Factor GenePrpf31Implicated in Human Autosomal Dominant Retinitis Pigmentosa (RP)". Investigative Opthalmology & Visual Science. 50 (12): 5927. doi:10.1167/iovs.08-3275. ISSN 1552-5783.
- ^ a b https://web.archive.org/web/20170329143940/http://kellogg.umich.edu/patientcare/downloads/Understand-Retinitis-Pigmentosa.pdf
- ^ https://pubmed.ncbi.nlm.nih.gov/20301590
- ^ Chang, Susie; Vaccarella, Leah; Olatunji, Sunday; Cebulla, Colleen; Christoforidis, John (2011-06). "Diagnostic Challenges in Retinitis Pigmentosa: Genotypic Multiplicity and Phenotypic Variability". Current Genomics. 12 (4): 267–275. doi:10.2174/138920211795860116. ISSN 1389-2029. PMC 3131734 $2. PMID 22131872. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ "Retinitis Pigmentosa". ColumbiaDoctors (İngilizce). 26 Nisan 2022. Erişim tarihi: 26 Temmuz 2024.
- ^ Schwartz, Stephen G; Wang, Xue; Chavis, Pamela; Kuriyan, Ajay E; Abariga, Samuel A (23 Haziran 2020). "Vitamin A and fish oils for preventing the progression of retinitis pigmentosa". The Cochrane Database of Systematic Reviews. 2020 (6): CD008428. doi:10.1002/14651858.CD008428.pub3. ISSN 1469-493X. PMC 7388842 $2. PMID 32573764.
- ^ a b Lok, Corie (1 Eylül 2014). "Curing blindness: Vision quest". Nature (İngilizce). 513 (7517): 160–162. doi:10.1038/513160a. ISSN 1476-4687.
- ^ jamanetwork.com. doi:10.1001/archopht.1993.01090060049022 https://jamanetwork.com/journals/jamaophthalmology/fullarticle/640248. Erişim tarihi: 26 Temmuz 2024. Eksik ya da boş
|başlık=(yardım) - ^ Berson, Eliot L. (2007-07). "Long-Term Visual Prognoses in Patients with Retinitis Pigmentosa The Ludwig von Sallmann Lecture". Experimental eye research. 85 (1): 7–14. doi:10.1016/j.exer.2007.03.001. ISSN 0014-4835. PMC 2892386 $2. PMID 17531222. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ "ClinicalTrials.gov". www.clinicaltrials.gov. Erişim tarihi: 26 Temmuz 2024.
- ^ Weiss, Jeffrey N.; Levy, Steven (2018-06). "Stem Cell Ophthalmology Treatment Study: bone marrow derived stem cells in the treatment of Retinitis Pigmentosa". Stem Cell Investigation. 5: 18–18. doi:10.21037/sci.2018.04.02. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ "How Does Argus® II Produce Sight?". archive.ph. 19 Ağustos 2013. Erişim tarihi: 26 Temmuz 2024.
- ^ Humayun, Mark S.; Dorn, Jessy D.; da Cruz, Lyndon; Dagnelie, Gislin; Sahel, José-Alain; Stanga, Paulo E.; Cideciyan, Artur V.; Duncan, Jacque L.; Eliott, Dean; Filley, Eugene; Ho, Allen C. (2012-04). "Interim Results from the International Trial of Second Sight's Visual Prosthesis". Ophthalmology. 119 (4): 779–788. doi:10.1016/j.ophtha.2011.09.028. ISSN 0161-6420. PMC 3319859 $2. PMID 22244176. Tarih değerini gözden geçirin:
|tarih=(yardım)KB1 bakım: PMC biçimi (link) - ^ "FDA approves first retinal implant for adults with rare genetic eye disease". web.archive.org. 16 Şubat 2013. Erişim tarihi: 26 Temmuz 2024.
- ^ "'First bionic eye' retinal chip for blind". ScienceDaily (İngilizce). Erişim tarihi: 26 Temmuz 2024.
- ^ Stingl, Katarina; Bartz-Schmidt, Karl Ulrich; Besch, Dorothea; Braun, Angelika; Bruckmann, Anna; Gekeler, Florian; Greppmaier, Udo; Hipp, Stephanie; Hörtdörfer, Gernot; Kernstock, Christoph; Koitschev, Assen (22 Nisan 2013). "Artificial vision with wirelessly powered subretinal electronic implant alpha-IMS". Proceedings of the Royal Society B: Biological Sciences. 280 (1757): 20130077. doi:10.1098/rspb.2013.0077. ISSN 0962-8452. PMC 3619489 $2. PMID 23427175.
- ^ Bainbridge, James W.B.; Smith, Alexander J.; Barker, Susie S.; Robbie, Scott; Henderson, Robert; Balaggan, Kamaljit; Viswanathan, Ananth; Holder, Graham E.; Stockman, Andrew; Tyler, Nick; Petersen-Jones, Simon (22 Mayıs 2008). "Effect of Gene Therapy on Visual Function in Leber's Congenital Amaurosis". New England Journal of Medicine (İngilizce). 358 (21): 2231–2239. doi:10.1056/NEJMoa0802268. ISSN 0028-4793.
- ^ Maguire, Albert M; High, Katherine A; Auricchio, Alberto; Wright, J Fraser; Pierce, Eric A; Testa, Francesco; Mingozzi, Federico; Bennicelli, Jeannette L; Ying, Gui-shuang; Rossi, Settimio; Fulton, Ann (7 Kasım 2009). "Age-dependent effects of RPE65 gene therapy for Leber's congenital amaurosis: a phase 1 dose-escalation trial". Lancet (London, England). 374 (9701): 1597–1605. doi:10.1016/S0140-6736(09)61836-5. ISSN 0140-6736. PMC 4492302 $2. PMID 19854499.
- ^ https://www.urmc.rochester.edu/news/publications/neuroscience/a-key-to-restoring-sight-may-be-held-in-a-drug-that-treats-alcoholism
- ^ Telias, Michael; Sit, Kevin K.; Frozenfar, Daniel; Smith, Benjamin; Misra, Arjit; Goard, Michael J.; Kramer, Richard H. "Retinoic acid inhibitors mitigate vision loss in a mouse model of retinal degeneration". Science Advances. 8 (11): eabm4643. doi:10.1126/sciadv.abm4643. ISSN 2375-2548. PMC 8932665 $2. PMID 35302843.
- ^ Papadopoulos, Loukia. "A drug once used to treat alcoholism may cure retinal degeneration". Interesting Engineering (İngilizce). Erişim tarihi: 26 Temmuz 2024.
- ^ Mezer, Eedy; Babul-Hirji, Riyana; Wise, Richard; Chipman, Mary; DaSilva, Lisa; Rowell, Mary; Thackray, Robin; Shuman, Cheryl T.; Levin, Alex V. (2007-01). "Attitudes Regarding Predictive Testing for Retinitis Pigmentosa". Ophthalmic Genetics (İngilizce). 28 (1): 9–15. doi:10.1080/13816810701199423. ISSN 1381-6810. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ Parmeggiani, Francesco (2011-06). "Clinics, Epidemiology and Genetics of Retinitis Pigmentosa". Current Genomics. 12 (4): 236–237. doi:10.2174/138920211795860080. ISSN 1389-2029. PMC 3131730 $2. PMID 22131868. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ Hamel, Christian (11 Ekim 2006). "Retinitis pigmentosa". Orphanet Journal of Rare Diseases. 1: 40. doi:10.1186/1750-1172-1-40. ISSN 1750-1172. PMC 1621055 $2. PMID 17032466.
- ^ Prokisch, Holger; Hartig, Monika; Hellinger, Rosa; Meitinger, Thomas; Rosenberg, Thomas (1 Eylül 2007). "A Population-Based Epidemiological and Genetic Study of X-Linked Retinitis Pigmentosa". Investigative Opthalmology & Visual Science. 48 (9): 4012. doi:10.1167/iovs.07-0071. ISSN 1552-5783.
- ^ http://onlinelibrary.wiley.com/store/10.1046/j.1395-3907.2002.00001.x/asset/j.1395-3907.2002.00001.x.pdf;jsessionid=DFAE926129B842C779D7F3FF4161710F.f02t01?v=1&t=i9kqhu90&s=5d78760662660ed7e8d11c378c2c396095ea2017
- ^ Graham-Rowe, Duncan (8 Eylül 2008). "Retinal transplants see fleeting success". Nature (İngilizce). doi:10.1038/news.2008.1088. ISSN 1476-4687.
- ^ "News Releases". web.archive.org. 8 Şubat 2005. Erişim tarihi: 26 Temmuz 2024.
- ^ http://iovs.arvojournals.org/article.aspx?articleid=2353744
- ^ "Error". www.abstractsonline.com. Erişim tarihi: 26 Temmuz 2024.
- ^ Tochitsky, Ivan; Polosukhina, Aleksandra; Degtyar, Vadim E.; Gallerani, Nicholas; Smith, Caleb M.; Friedman, Aaron; Van Gelder, Russell N.; Trauner, Dirk; Kaufer, Daniela; Kramer, Richard H. (19 Şubat 2014). "Restoring visual function to blind mice with a photoswitch that exploits electrophysiological remodeling of retinal ganglion cells". Neuron. 81 (4): 800–813. doi:10.1016/j.neuron.2014.01.003. ISSN 0896-6273. PMC 3933823 $2. PMID 24559673.
- ^ Bakondi, Benjamin; Lv, Wenjian; Lu, Bin; Jones, Melissa K; Tsai, Yuchun; Kim, Kevin J; Levy, Rachelle; Akhtar, Aslam Abbasi; Breunig, Joshua J; Svendsen, Clive N; Wang, Shaomei (2016-03). "In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa". Molecular Therapy. 24 (3): 556–563. doi:10.1038/mt.2015.220. ISSN 1525-0016. PMC 4786918 $2. PMID 26666451. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ Byrne, Leah C.; Dalkara, Deniz; Luna, Gabriel; Fisher, Steven K.; Clérin, Emmanuelle; Sahel, Jose-Alain; Léveillard, Thierry; Flannery, John G. (2 Ocak 2015). "Viral-mediated RdCVF and RdCVFL expression protects cone and rod photoreceptors in retinal degeneration". The Journal of Clinical Investigation (İngilizce). 125 (1): 105–116. doi:10.1172/JCI65654. ISSN 0021-9738. PMC 4382269 $2. PMID 25415434. KB1 bakım: PMC biçimi (link)
- ^ Xiong, Wenjun; MacColl Garfinkel, Alexandra E.; Li, Yiqing; Benowitz, Larry I.; Cepko, Constance L. (1 Nisan 2015). "NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage". The Journal of Clinical Investigation. 125 (4): 1433–1445. doi:10.1172/JCI79735. ISSN 0021-9738. PMC 4396467 $2. PMID 25798616.
- ^ a b Commissioner, Office of the (24 Mart 2020). "FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss". FDA (İngilizce). Erişim tarihi: 26 Temmuz 2024.
- ^ "Texas Woman Is the First Person to Undergo Optogenetic Therapy". MIT Technology Review (İngilizce). Erişim tarihi: 26 Temmuz 2024.
- ^ Sahel, José-Alain; Boulanger-Scemama, Elise; Pagot, Chloé; Arleo, Angelo; Galluppi, Francesco; Martel, Joseph N.; Esposti, Simona Degli; Delaux, Alexandre; de Saint Aubert, Jean-Baptiste; de Montleau, Caroline; Gutman, Emmanuel (2021-07). "Partial recovery of visual function in a blind patient after optogenetic therapy". Nature Medicine (İngilizce). 27 (7): 1223–1229. doi:10.1038/s41591-021-01351-4. ISSN 1546-170X. Tarih değerini gözden geçirin:
|tarih=(yardım) - ^ "Algae proteins partially restore man's sight" (İngilizce). 24 Mayıs 2021. Erişim tarihi: 26 Temmuz 2024.
- ^ Critic, Carly Maga Theatre (12 Aralık 2017). "Blind actor Alex Bulmer leads the way into theatre's future". Toronto Star (İngilizce). Erişim tarihi: 26 Temmuz 2024.
- ^ correspondent, Lauren Daley Globe; September 29, Updated; 2022; Comments, 12:42 p m Share on Facebook Share on TwitterView. "Losing his vision has opened Mark Erelli's eyes - The Boston Globe". BostonGlobe.com (İngilizce). Erişim tarihi: 26 Temmuz 2024.
- ^ "Neil Fachie - British Paralympic Association". web.archive.org. 27 Haziran 2012. Erişim tarihi: 26 Temmuz 2024.
- ^ http://www.theaustralian.com.au/news/proud-lindy-rides-for-two-countries/story-e6frg7mo-1111116493868
- ^ Star-Ledger, Salvador Rizzo | The (25 Eylül 2013). "Lonegan opens up about his blindness". nj (İngilizce). Erişim tarihi: 26 Temmuz 2024.
- ^ Thomson, Alice (26 Temmuz 2024). "'I was 16. My doctor said, "You'll go blind. Get on with it" '". www.thetimes.com (İngilizce). Erişim tarihi: 26 Temmuz 2024.
- ^ Spencer, Frederick J. (2002). Jazz and death : medical profiles of jazz greats. The Archive of Contemporary Music. Jackson : University Press of Mississippi. ISBN 978-1-57806-453-3.
- ^ https://en.wikipedia.org/wiki/Retinitis_pigmentosa#cite_note-76
- ^ "Shel Talmy Part Two". spectropop.com. Erişim tarihi: 26 Temmuz 2024.
- ^ "Danelle Umstead". web.archive.org. 1 Mayıs 2015. Erişim tarihi: 26 Temmuz 2024.
- ^ http://www.cbs.com/primetime/csi/cast/jon-wellner/
- ^ Paumgarten, Nick (15 Ekim 2006). "The $40-Million Elbow". The New Yorker (İngilizce). ISSN 0028-792X. Erişim tarihi: 26 Temmuz 2024.
- ^ "Take 5: Sheena Iyengar, author and expert on choice | NJ.com". web.archive.org. 10 Mayıs 2018. Erişim tarihi: 26 Temmuz 2024.
Dış bağlantılar
 | Göz ile ilgili bu madde taslak seviyesindedir. Madde içeriğini genişleterek Vikipedi'ye katkı sağlayabilirsiniz. |
 | Hastalık ile ilgili bu madde taslak seviyesindedir. Madde içeriğini genişleterek Vikipedi'ye katkı sağlayabilirsiniz. |
 Göz hastalıkları Göz hastalıkları | |
|---|---|
| Sınıflandırma | D |
|---|---|
| Dış kaynaklar |
|









